

R数据分析:孟德尔随机化实操 - Codewar
source link: https://www.cnblogs.com/Codewar/p/16881800.html
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

R数据分析:孟德尔随机化实操
好多同学询问孟德尔随机化的问题,我再来尝试着梳理一遍,希望对大家有所帮助,首先看下图1分钟,盯着看将下图印在脑海中:
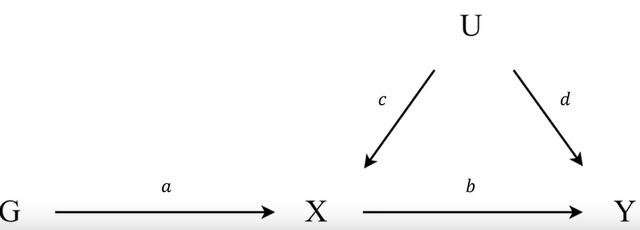
上图是工具变量(不知道工具变量请翻之前的文章)的模式图,明确一个点:我们做孟德尔的时候感兴趣的是x和y的关系,也就是小b,但是我们直接去跑x对y的回归肯定是不对的,因为有很多的U,因此我们借助工具变量G(关于工具变量我们之前的文章有详细的解释,请自行查阅),去估计我们感兴趣的小b。
现在有天然良好的工具变量G,也就是我们的基因变量,此时有上面的图,再次重申:我们感兴趣的,最终希望得到准确估计的值是小b,按照上图我们应该有GY的关系是ab,GX的关系是a,于是乎b可以写成ab/a,就是我们感兴趣的b可以换一种思路得到,如下:

上面的式子要跑通的话,我们需要知道G-Y的关系和G-X的关系。
但是我们GY也就是基因和结局的关系已经有人给我们研究好了,我们可以直接去GWAS里面找研究好的summarydata拿来用就行。
但是我们的的GX也就是基因和暴露的关系也已经有人给我们研究好了,我们可以直接去GWAS里面找研究好的summarydata拿来用就行。
也就是说,通过孟德尔随机化,我们完全可以毫不费力地估计出我们需要的小b,也就是暴露和结局的关系----就是今天要再次给大家介绍的孟德尔随机化研究。
思路就是这么清晰。就是这么清晰。搞不明白的同学再多读几遍。
为了帮助大家理解思想,在孟德尔随机化的实操中有几个术语得提点一波:
连锁不平衡(linkage disequilibrium):刚刚讲我们可以有很多的基因结局/暴露的关系的,就是GWAS里面好些基因可以用,这个时候我们不希望基因之间有相关(会造成double counting,使得结果偏倚):
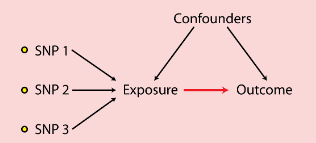
我们实际做的时候,模式是像上图,snp之间你说不相干就不相干?当两个位点的不同等位基因的关联频率高于或低于独立随机关联的条件下的期望频率,这种情况是客观存在的,此时时这些工具变量之间相关性就叫连锁不平衡,其大小可以用LD r方来表示,这个指标也是我们在操作时需要设定的指标之一。
水平基因多效性(Horizontal Pleiotropy):理解这个概念先看下图:
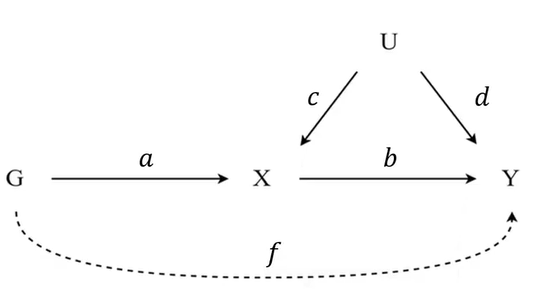
意思是我的理想的情况是通过ab/a的操作估计出b,但是看上图,是不是免不了会出现f这条路径,如果出现了f,我们的基因和结局之间的关系就是f+ab,此时,我用原来的方法估计的就不是b了,而是b+f/a了,就不对了(始终记住我们关心的是b)。
但是如果我的基因变量很多,从而有很多的f,如果所有f的期望均值为0,那么最后我们汇总一下得到的结果也基本上就是b了,无伤大雅。但是就怕所有的f都是一边偏向的(都大于0或都小于0),此时就有问题了,叫做定向多效性directional pleiotropy,这也是为什么我们最后要做漏斗图的原因。
就是通过漏斗图一看都是所有的工具变量都是呈漏斗分布的,就说明没有偏向,这个时候我们认为定向多效性都被冲掉了,不影响。
好,解释了上面的一些术语之后,我们实操一波。
最基本的例子:BMI on CHD的例子,我想看一下BMI作为暴露,CHD作为结局的mr,代码就4条:
bmi_exp_dat <- extract_instruments(outcomes = 'ieu-a-2')
chd_out_dat <- extract_outcome_data(snps = bmi_exp_dat$SNP, outcomes = 'ieu-a-7')
dat <- harmonise_data(bmi_exp_dat, chd_out_dat)
res <- mr(dat)结果如下,下图中有不同方法出来的我们关心的小b:
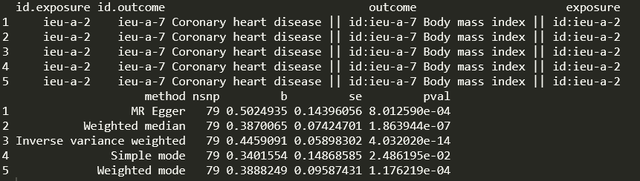
这个就算做完了,就这么简单快速。
接下来就是敏感性分析,首先是各个工具变量的异质性检验:
mr_heterogeneity(dat)运行代码后可以得到Cochran’s Q统计量
然后是水平基因多效性检验,代码如下:
mr_pleiotropy_test(dat)运行代码可以得到egger_intercept
然后是单个SNP结果检验,代码如下:
res_single <- mr_singlesnp(dat)运行后可以得到每个SNP的小b
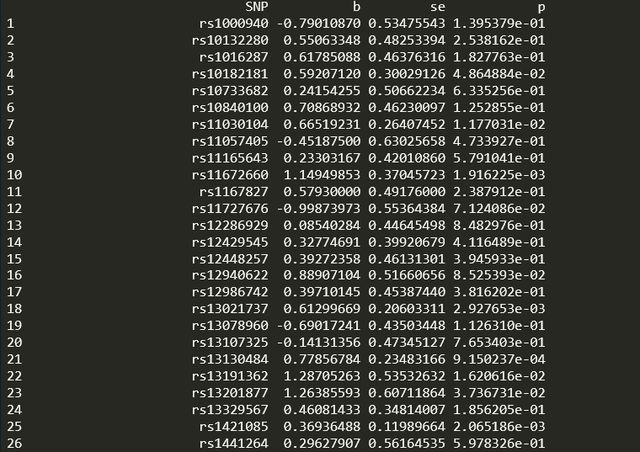
然后是留一检验,代码如下:
mr_leaveoneout(dat)接下来,论文中还会有几个图,首先是点图,代码如下:
mr_scatter_plot(res, dat)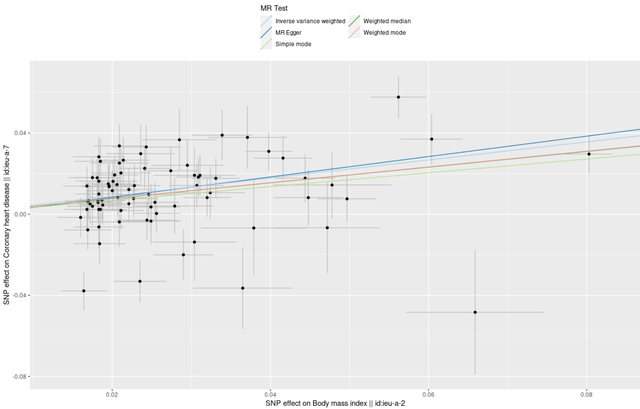
点图是将同一个SNP对暴露的效果放在横轴,对结局的效果放在纵轴,此时图中的斜率就是我们的估计的小b。
然后是单个SNP效应组合的森林图用mr_forest_plot函数可以得到,mr_leaveoneout_plot可以得到留一分析的森林图,mr_funnel_plot可以帮我们得到漏斗图。
到这就出了所有需要报告的东西,做完了。
但是上面的流程有很多的前提,比如你得知道暴露和结局的GWASid才能进行下去,GWAS又有很多,比如你直接用上面的代码的话其实是MR Base GWAS catalog里面的GWAS,当然你还可以选别的,或者用自己找来的最新的GWAS都是可以的。

第一步首先是在相应的GWAS中找到暴露的summary data:
那么有那些GWAS可以供我们使用呢?我们可以直接把GWAS的目录调出来瞅瞅,代码如下:
data(gwas_catalog)运行后大约可以得到15万个全基因组关联研究的数据,截图如下:
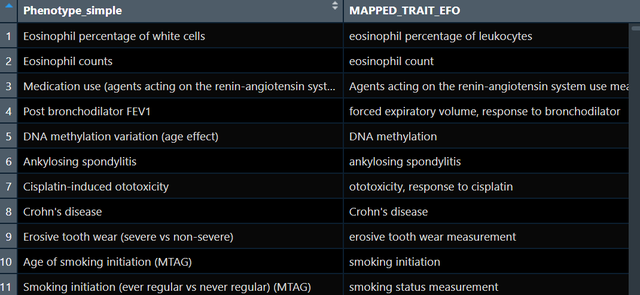
那么对我们而言,我们现在需要找到我们关心的暴露对应的GWAS,比如我现在要找与“blood”表型相关的GWAS,我可以写出如下代码:
exposure_gwas <- subset(gwas_catalog, grepl("Blood", Phenotype_simple))上面的代码相当于只用Phenotype_simple这一列做筛选,当然你也可以结合其它的列比如人群,比如作者,比如地区等等,都是可以的。
选好暴露相关的GWAS之后要做的就是进一步确定基因工具变量和暴露的强度,在论文中一般是这么描述:First, relevance assumption was met considering that all SNPs have reached genome-wide significance (p < 5 × 10−8)
具体的操作如下:
exposure_gwas<-exposure_gwas[exposure_gwas$pval<5*10^-8,]通过上面的步骤保证我们的基因工具变量一定是和暴露强相关。
然后就是将准备好的暴露的GWAS数据形成可以用来做MR分析的数据格式,需要用到format_data()函数:
exposure_data<-format_data(exposure_gwas)此时的exposure_data大概长这样:
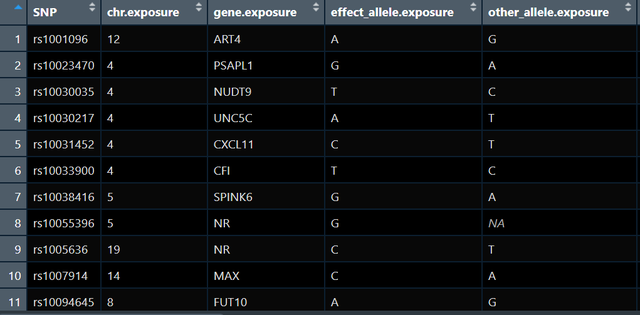
可以看到有很多个基因工具变量SNP,这个时候我们需要考虑连锁不平衡(linkage disequilibrium):
exposure_data<-clump_data(exposure_data, clump_r2 = 0.001)上面的代码中clump_r2则是设定的容许相关性,到这儿我们算是手动地将工具变量都筛出来了,解决了找工具变量的问题,还有一个方法是自动筛选工具变量,比如我暴露是bmi,我可以写出如下代码:
subset(ao, grepl("body mass", trait))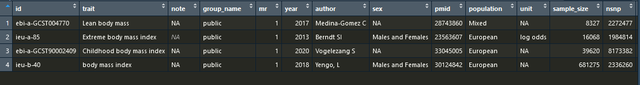
运行后我知道我可以选的gwasid是ieu-b-40,这个时候我也可以自动提取出工具变量,这两种方法达到的目的都是一样的:
extract_instruments('ieu-b-40')然后依照工具变量进行结局的summary estimates的提取,提取结局的summary data也需要是需要知道GWASid的对吧,比如我现在关心的结局是收缩压,我就可以写出如下代码:
outcome_gwas <- subset(ao, grepl("Systolic", trait))运行后我就可以知道所有的和收缩压相关的gwasid了,我选一个最新的,比如我就选下面的2021年的:
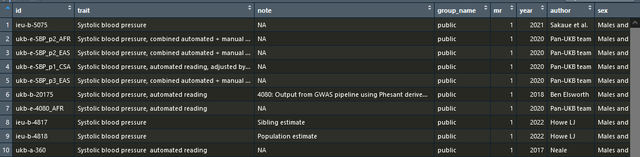
看图我们知道它对于的id是ieu-b-5075,我就这么写:
outcome_data <- extract_outcome_data(
snps = exposure_data$SNP, outcomes = "ieu-b-5075")后续通过合并直接做mr分析就可以,流程就没有不同了。
今天给大家写了孟德尔随机话的实操,文章图示例来自【中文孟德尔随机化】英国布里斯托大学MRC-IEU《R语言做孟德尔随机化》第一章:用MRBase网页工具和R包TwoSampleMR做两样本孟德尔随机化_哔哩哔哩_bilibili,感谢大家耐心看完
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK