

深度学习应用系列——DeepVariant:鉴定遗传变异
source link: https://szjshuffle.github.io/2019/08-08-deepvariant.html
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

DeepVariant
A universal SNP and small-indel variant caller using deep neural networks
DeepVariant是由Google Brain Genomics团队提出的一种基于CNN的DNA遗传变异识别算法,在PrecisionFDA TruthChallenge比赛中取得了最优奖,并于18年发表在nature子刊nature biotechnology上。
DeepVariant首先基于高灵敏度的生物信息学方法找到测序数据中可能存在突变的候选位点,随后将这些位点的pileup(可以理解为比对结果)编码成7个channel的图像,并送入预训练过的Google Inception V2模型中,通过softmax函数输出候选位点对应的三种不同基因型的概率。最后再对候选位点进行合并处理,输出最终的突变检测VCF文件。
测序技术在过去二十年内取得了快速进步,但由于测序误差以及个体基因组不尽相同的突变情况,准确地从测序得到的百万条序列片段(reads)中识别个体基因组中的遗传变异仍然非常具有挑战性。遗传变异的识别是精准医疗的基石,因此开发一种准确识别遗传变异的算法至关重要。
目前,行业内的主流软件是GATK(Genome Analysis Toolkit),它基于传统的统计学习理论,同时依赖专家手动设计特征(首先运行逻辑回归来去除测序错误,然后用HMM计算读段似然,朴素贝叶斯方法识别遗传突变,最后用GMM去除低质量的突变。 ),取得很好的效果。但唯一的缺陷在于无法兼容很好的不同测序平台得到的数据。而深度学习的引入使得流程更加自动化,突变识别准确率更高,并且能够在不同测序平台中使用。
本文主要工作介绍
DeepVariant的工作流程如图所示。
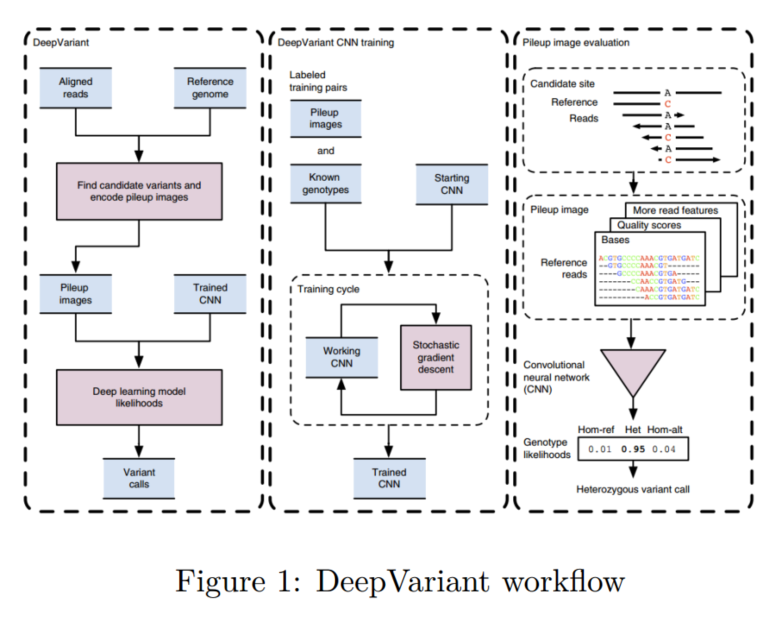
其大致可以分成以下几步:
1.生成样本
序列比对到参考基因组上,利用生物信息学方法识别潜在的突变位点。具体过程:比对完毕后会得到一个VCF文件(Variant Calling Format),将BAM文件(bam文件是比对过程中产生的一个文件,用于记录比对结果信息)中的CIGAR string解析出来,从而得到一些潜在的突变位点,并将其pileup图片保存下来(pileup可以用基因组浏览器查看)。pileup图片大致如下所示:
.png)
接下来在一个pileup图像中,根据reads提供的突变个数生成若干张图像。比如在上图中,正中间的位点参考基因组碱基为A,测序得到的reads显示该位点含有G和T两种,因此可能存在3种等位基因({G,T,G&T}),因此生成3张图像。并结合真实的突变信息(基因组数据集来自Genome In A Bottle (GIAB),这套数据集上的突变位点已经经过了实验验证,这就相当于训练数据被完美的标注过了。)进行label,一共有3种标签:
(1)homozygous reference:纯合子并且和参考基因组相同;
(2)heterozygous:杂合子;
(3)homozygous alternative:纯合子但与参考基因组不同。
2.编码样本
将pileup信息编码成6个通道作为DeepVarient的输入,第一步生成的每一个pileup图像就是一个训练样本,每个样本都被重新编码成了一张6个通道的tensor图像(221x100x6),作为DeepVarient的输入。
(由于全基因组测序数据的平均覆盖深度一般不会超过100,DeepVariant实际产生的图像高度设置成了100,对于测序覆盖度大于100的区域,则通过下采样技术将其覆盖度降低至100以满足图像生成的要求。同时现有主流测序平台产生的测序数据读长在100bp左右,对于候选的变异位点,向基因组两端延伸100bp即可将大部分覆盖待检位点的测序数据包含进来,考虑到插入缺失变异的长度,为其预留20bp的编码空间,进而图像宽度设置成了221,这样实际产生的图像大小是221x100)
这6个通道分别为:
a) read base(b) base quality, (c) mapping quality, (d) strand, (e) support variant, and (f) support reference.
一个例子如下图所示(其中第一张是后面6张的汇总):
.png)
3.训练网络
训练的过程本质就是建立tensor图像到基因型标签的映射关系。
网络选用Google Inception V2,网络结构如下图所示:
.png)
特点是通过堆叠多个的Inception模块,构建层次更深的网络结构,提高网络的判定能力,并且由于采用了较小尺寸的卷积核,从而降低了网络的参数数目,加快了网络的训练速度。
DeepVariant采用的CNN网络的输入层尺寸是299×299,因此需要对原始图像(221x100x6)进行缩放(rescaling),在不改变像素值的情况下将其尺寸放大到299×299。
网络初始化参数全部采用ImageNet中训练得到的参数。
网络优化方法:mini-batch GD(batch = 32) & RMS decay = 0.9;
4.生成VCF文件
把训练好的网络参数保存后,对于unlabeled样本,将其输入到网络后,ke’yi’de
评估算法效果
在PrecisionFDA TruthChallenge比赛提供的数据集中,通过与主流的软件进行对比,部分结果如图:
.png)
DeepVariant的错误识别数小于第二好的算法50%。
一些个人的思考
尽管DeepVariant在突变识别上取得了很大的突破,但是受到其网络输入尺寸的限制,没有办法识别读长较大的测序数据,例如:单分子测序数据。
近两年,更多基于深度学习的变异检测算法也开始出现。例如,VariantNET和Clairvoyante是使用多任务学习设计的,用于噪声较大的单分子DNA测序数据。Clairvoyante训练深度神经网络,以较小的网络同时预测变异类型(het或hom,SNP或indels)和等位基因。
相信深度学习能为基因组学带来更多的突破。
[1] Poplin, Ryan; Chang, Pi-Chuan; Alexander, David; Schwartz, Scott; Colthurst, Thomas; Ku, Alexander; Newburger, Dan; Dijamco, Jojo; Nguyen, Nam; Afshar, Pegah T; Gross, Sam S; Dorfman, Lizzie; McLean, Cory Y; DePristo, Mark A. “A universal SNP and small-indel variant caller using deep neural networks”. In: Nature Biotechnology (2018). issn: 1087-0156,1546-1696. doi: 10.1038/nbt.4235.
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK