

详解DADA2算法原理
source link: https://szjshuffle.github.io/2019/11-02-DADA2%E9%98%85%E8%AF%BB%E7%AC%94%E8%AE%B0.html
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

DADA2
题目:dada2: high-resolution sample inference from illumina amplicon datas
作者:Benjamin J Callahan1, Paul J McMurdie,Michael J Rosen, Andrew W Han, Amy Jo A Johnson2 & Susan P Holmes1
杂志:Nature methods, published: 23 MAy,2016
DADA2是一个用于建模和纠正Illumina扩增子测序错误的算法(https://github.com/benjjneb/dada2)。
DADA2准确地推断出样品序列,并解决了仅1个核苷酸的差异问题。
在一些模拟社区中,与其他方法相比,DADA2识别出更多的真实变异并输出了更少的伪序列。
扩增子测序中一个很重要的问题是如何从在带有测序错误的情况下(很显然,基于OTU的方法没有考虑测序错误的因素),从而解析真正的生物学变异。该挑战促使人们开发了扩增子的错误校正方法。
在DADA2发表以前,尚无针对illumina平台的错误校正方法。
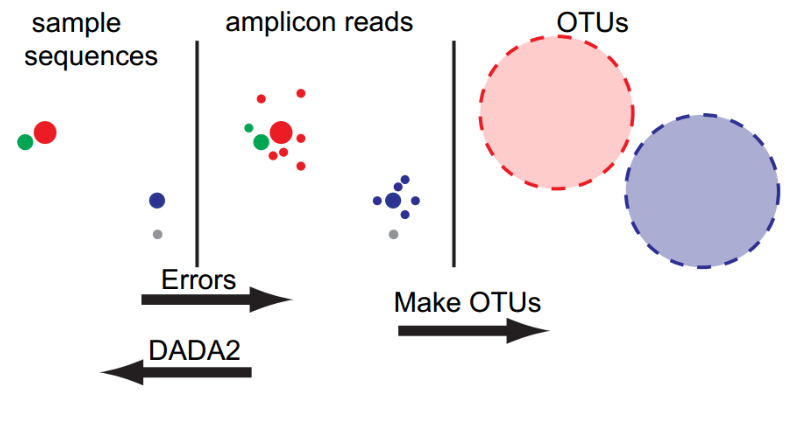
- otu方法的两大致命问题
目前,基于illumina平台扩增子测序错误的解决办主要是通过:先质量过滤,随后构建OTU来解决。
1.简单的根据相似度进行聚类会错把测序错误当做生物学变异,因此OTU的方法并没有进行假阳性推断。
2.此外,OTU方法没有利用测序错误模型,从而无法进行精细尺度变异(Fine-scale variation)的解析。
- 精细尺度变异与DADA2
精细尺度变异(Fine-scale variation)的好处很多。最浅显的:可以将某些菌株中的致病菌与普通菌株区分开,并且可以在复杂的微生物组相关疾病中提供更多的临床相关信息。
DADA2完整的扩增子工作流程:过滤,重复数据删除,样本推论,嵌合体识别以及成对末端读段的合并。
1.根据error model计算error rate,error rate是量化了测序序列i产生自样本序列j的概率,error rate是序列碱基组成以及碱基质量的函数。
2.理论上,read i产生自read j的数量可以描述为泊松分布,期望值lambda= error rate x (# of read j)。如果i真的来自j,那么观测值(即,i的丰度)应当与期望值相等。根据这一事实,可以进行假设检验。计算的p-value记作abundance p-value.
3.abundance p-value被用于作为迭代partition算法的是否要继续迭代的阈值标准;当所有abundance p-value均满足阈值要求时,我们可以认为:同一个partition下的序列都产生于该partition的central sequence。最终,推算出来的sample组成就是每一个Partition中的center 序列以及对应的丰度。
【注:一个partition可以看做一个cluster,降噪体现在最后是以center序列作为partition的代表,这样就消除了partition内的其他序列由于测序原因而引入的错误。】
error model
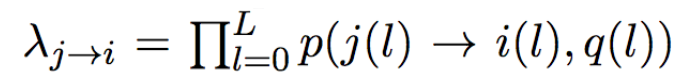
dada2假设在reads内的测序错误以及reads之间是相互独立的。
在这种假设下,样本真实的序列j产生测序序列i的错误率(error rate,即公式中的Lambda)可以描述为:每一个碱基替换概率的累乘函数。此函数与替换的碱基类型(也包含不发生替换的情况),碱基测序质量得分有关。
例如,p(A→C, 35) 表示该碱基测序得分是35分的情况下,从A替换为C的概率(显然是一个很小的值)。
当然,在实际算法运行过程中,显然在起始条件下并不知道哪一条序列是真实的样本序列j,因此,采用启发式的策略,人为的把在一个partition下丰度最高的unique read假定为真实序列j,其他unique read与j进行比对,并计算相应的lambda(j->i),(具体参考partition一节)。
如果序列j和序列i很相似,error rate应该是一个很接近1的值。如果两者匹配不上,那就直接把error rate分配为0。
abundance p-value
如果测序的错误在一段reads上是独立的,那么,真实的序列j产生测序序列i的数量应当服从泊松分布,其中测序序列i的期望值应当为:
期望值=error rate x 期望的样本真实序列j数量
error rate可以从error model计算得到
期望的样本
真实序列j数量显然无法得到,但可以采用启发式算法进行迭代求解。(参考下一节)
根据这个期望值以及实际得到的观测值,我们就可以利用假设检验判断read i是否来自于序列j。
因此,进一步进行假设检验:
H0:测序序列i的观测丰度 = 理论期望值
H1:两者不相等
p-value可以用于衡量一条测序序列i产生自真实的样本序列j的可能性大小。
如果P-value很大,则无法拒绝原假设,我们就认为测序序列i是来自序列j,即i和j最后被划分为同一个partition;
计算p-value:
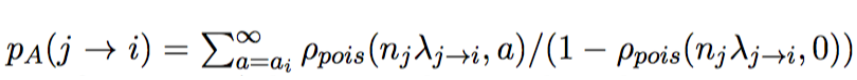 其中:
其中:
j是假设的来自样本的真实序列,i是测序序列,n_j是期望的样本真实序列j数量,lamda是error rate,a_i是read i的实际观测值。
n_jxlambda就是真实的序列j产生测序序列i的数量的期望值。
个人的思考:
p-value的定义:the probability we get this sample and a more extreme sample under H0.
因此,根据p-value的定义:在本例子下,我们的p-value可以计算为:序列i的丰度观测值a_i以及更多的情况下的概率。
所以,原论文中Then, conditional on i being read at least once, the abundance pvalue is the probability of seeing nj or more identical reads 的表述应该是错误的。
公式中的分母:
可以这么解释:
除以1- p(0)是为了剔除掉观测值为0的情况(显然,观测值不可能为0)。由于概率分布之和为1,因此除以1-p(0)进行重新归一化。
显然,p-value越小,越表明现有的reads丰度无法被error model所解释,也就意味着这个测序序列i不是来自于该真实序列j(再一次强调,在实际算法运行过程中,显然在起始条件下并不知道哪一条序列是真实的样本序列j,因此,采用启发式的策略,人为的把丰度最高的unique read假定为真实序列j,其他unique read与j进行比对,并计算error rate 以及进一步根据error rate计算abundance p-value)
1.p-value显然需要一个阈值,用于判断是否显著,这个值通过OMEGA_A 参数进行设定,默认:1e-40。
2.启发式算法采用迭代策略,直到所有partition内的abundance p-value都大于阈值。
3.singleton 的p-value被设定为1,并且不会被error model判断与真实序列j的一致性。这意味着,singleton无法构建partition(具体参考partition:分配算法一节),因此DADA2不会推断singleton
partition:分配算法
前面一直提到:在实际算法运行过程中,显然在起始条件下并不知道哪一条序列是真实的样本序列j,因此,采用启发式的策略,人为的把丰度最高的unique read假定为真实序列j,其他unique read与j进行比对,并计算error rate 以及进一步根据error rate计算abundance p-value)。
具体的分配步骤:
1.扩增序列被进行去冗余,从而得到若干unique reads;每一个unique read可以根据原始reads数量换算成丰度,以及求取平均质量。(平均质量需要被用于error model)
2.将所有unique reads分配到一个partition中,将丰度最高的read分配为中心。
人为将丰度最高的reads作为中心,并假设为真正来自于样本序列,个人认为这是因为:
由于聚类过程是根据完全一致性进行unique reads划分,这个过程很显然,由于测序的错误,许多本是来自于同一个样本序列的reads会被划分为不同的unique reads;而丰度最高的unique reads显然可信度是最高的,因此也可以认为这个unique reads与样本真实序列是最近的。
其他所有unique reads与中心read进行比对,计算error rate: lambda(center->each unique read),以及进一步计算abundance p-value。
3.如果最小的p-value(经过bonferroni校正)小于用户设定的阈值OMEGA_A (根据经验设定,默认是),这就表明算法认为该partition中仍然存在着不是来自于真实序列(partition center)的测序reads;此时,将继续创建一个新的partition,并把p-value最小的reads分配为新的中心,其他所有unique reads与新的中心逐一进行比对,一旦发现新的中心更可能产生该unique reads,那么就把该unique reads划分为新的partition。
4.第3步不断进行迭代,直到所有的p-value都大于OMEGA_A 。此时,可以认为,每条序列已经被划分为最可能产生它的partition中。
最终,根据1-4步推算出来的sample组成就是每一个Partition中的center 序列以及对应的丰度。(降噪体现在最后是以center序列作为partition的代表,partition内的其他序列由于测序错误的原因,最后直接被忽略了)
error model的参数
前面提到的error model中的替换概率问题,error rate的计算依赖于每一个碱基替换的概率,而每一个碱基的替换情况由于实际碱基测序质量有关。
因此,实际上存在16x41个参数
41代表了所有质量得分情况(1~41),16是4种碱基替换情况,见下图:
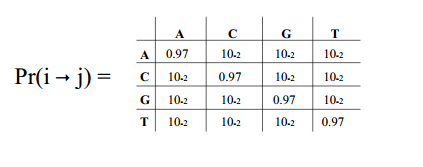
因此DADA2提供了3种选择:
1.基于用户的实际数据进行参数估计(利用dada2内置算法selfConsist )
2.(默认)单独对每一种替换,进行基于weighted loess 的拟合
regularized log (observed mismatch rates ) ~ weighted loess function(mismatch base quality)
3.用户自己定义一个参数估计函数。
- benchmark
与4种算法进行了比较:
UPARSE
mothur(平均链接)
QIIME(uclust)
在模拟社区进行了测评:Balanced,HMP和Extreme
平衡的社区包含在理论上相等的频率:57种细菌和古细菌。
HMP社区包含21个细菌在名义上相等的频率17,
而极端群落包含27个细菌菌株,其频率跨越五个数量级,在整个序列区域上相差至少1个核苷酸(nt)。
将输出序列与组成这些模拟社区的参考序列进行了比较,根据算法结果,输出序列被分成了3种:
Exact hit: blast比对完全比对上了参考序列。
One-off:存在一个Mismatch 或者 gap。
Other:其他情况。
sensitivity :定义为Exact hit的比例。(需要注意的是:由于精细尺度变异广泛存在于模拟社区中,因此有些参考的菌株在16s序列中包含了多个可以区分的序列变异。)
DADA2软件流程
分裂扩增子去噪算法(Divisive Amplicon Denoising Algorithm.)
考虑了测序质量,无需参考数据库。
核心降噪算法是基于Illumina测序错误模型。
1.过滤: fastqFilter()
与usearch fastq_filter命令类似
此函数将序列修剪为指定的长度,删除短于该长度的序列,并根据歧义碱基的数量、最低得分、reads中的期望错误进行过滤。
fastqPairedFilter() 实现了同样地功能,只不过输出的是前、后端都通过过滤的reads。
2.重复删除 :derepFastq()
derepFastq()导入一个fastq文件并输出重复序列的唯一序列及其丰度。
derepFastq()还会输出一致序列的碱基质量得分(通过计算一致序列中对应位置的碱基质量平均值。)
该质量得分用于error model的dada函数。
3.降噪:dada()
细节在前面已经描述。
4.去除嵌合体:isBimeraDenovo()
去除嵌合体最好dada()之后,推断样本系列之前进行。
而且,最好不要使用独立的嵌合体去除算法(因为当前大多数独立的嵌合体识别程序,在识别与其他更丰富序列时更加保守,因为此类序列有望在以后合并在同一OTU中)。
具体实现:
首先,嵌合体所来源的序列丰度一定要比嵌合体大。
因此,嵌合体序列是通过与丰度更高的序列进行Needleman-Wunsch 全局比对发现的,如果嵌合体的左端或者右端能与母序列毫无差别的匹配上,就认为是嵌合体。如果嵌合体序列与母序列存在单个碱基/indel的差别,也会被标记。
5.合并双端序列mergePairs()
如果两段序列确实存在overlapp,就merge起来。注意到在DADA2管道中合并是在去噪后,因此严格要求精确重叠(因为预几乎所有替代错误都已经已被降噪算法删除)。
1.降噪后再合并双端序列
这是因为核心去噪算法使用了质量得分和错误率之间的经验关系。如果序列合并过后,正向,重叠和反向序列部分的这种关系会变得不一致,从而干扰降噪算法。
补充1:精确序列变异缘何替代OTU?
用精确序列变异取代OTU,这样能够使标记基因测序:
1.更加有效精确(去噪算法),
2.可重复使用(即使更多的样本进来了也不需要重算),
3.可再现且全面(不同研究之间具有可比性)。
精确序列变异有很多种叫法,都是一个意思:
Exact Sequence Variants (ESVs)
- Callahan et al. 2017 (by accident)
Amplicon Sequence Variants (ASVs)
- Needham et al. 2017
- Callahan et al. 2017
sub-OTUs (sOTUs)
- Amir et al. 2017
Zero radius OTUs (zOTUs)
- Edgar 2017
Haplotypes, oligotypes, …
其中zOTU的叫法可以形象的用下图描述:

论文:dada2: high-resolution sample inference from illumina amplicon data
slides:https://www.biostat.washington.edu/sites/default/files/modules//2016-SISMID-14-01.pdf
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK