

基因组质量评估:(一)概述
source link: https://yanzhongsino.github.io/2022/07/10/omics_genome_quality.assessment_intro/
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

基因组质量评估:(一)概述
2022-07-10omics , genome , quality assessment
(全文约1600字)
1. 基因组质量评估
- 随着越来越多的组学测序数据的产生,基因组组装和注释变得越来越常见。
- 基因组组装和注释结果的质量评估对加强下游分析的科学性是非常有必要的。
2. 评估标准
Earth Biogenome Project(EBP)在2021年3月发布了4.0版本的核基因组组装质量标准报告。
可参考:https://www.earthbiogenome.org/assembly-standards。
2.1. 生物分组
评估标准把生物分成三组,对不同组的生物提出了不同的标准:
- 有足够DNA和材料可用的真核生物:6.C.Q40标准。
- 连续性(contiguity):contig N50和scaffold N50达到Mbp级别
- 错误率(error rate):小于1/10000的错误率
- <5% false duplications
- >90% kmer completeness
- >90%的序列可以align到候选染色体上
- >90%的单拷贝保守基因是完整且单拷贝的,例如用BUSCO评估
- 来自同一个组织的>90%转录本可以mapping到基因组
- DNA和材料有限的物种(例如单个个体的DNA<100ng):4.5.Q40标准
- 连续性(contiguity):contig N50和scaffold N50达到10kbp级别
- 错误率(error rate):小于1/10000的错误率
- 不可培养的单细胞真核生物
- 类似宏基因组学的标准,根据原核生物群落的经验来确定的。
2.2. 附加要求
由于自动化组装程序几乎都会在组装结果中保留一些错误,所以提出了一套质量控制标准,包括:
- 将污染源和其他物种(例如共生物种/寄生物种)的序列从目标物种的序列中分离去除。
- 识别初级组装(单倍型或假单倍型),应该是说尽量组装到染色体水平的意思。
- 分离去除和明确鉴定细胞器基因组。
- 只有A,C,G,T,N碱基,序列不应以Ns开头或结尾。
此外,还鼓励:
- 识别原始数据和生成的组装之间的不一致,以定位和消除结构错误(错误连接,丢失连接,错误重复)。
- 染色体的识别和命名,尤其是性染色体。
- 与已知的核型保持一致。
2.3. 提交要求
对于达到EBP要求的参考基因组,还需提交到公开数据库INSDC(GenBank/EMBL/DDBJ)以开放给科学同行。
- 提交到公开数据库的基因组需要连接到一个BioProject,并建议以下图的结构进行组织和维护。

Figure 1. BioProjects的层级结构 图源:https://www.earthbiogenome.org/assembly-standards
同时将基因组的物种分配到NCBI分类数据库的“txid”条目,与分类学上有效的物种进行关联。如果暂时不存在有效物种名,可以用例如*Maylandia sp. “pearly”*的txid标识符。
样本个体名称也应规范。EBP建议每个基因组样本的个体都应按照规范获取一个生命树ID(ToLID)。
- ToLID介绍:https://id.tol.sanger.ac.uk/
- ToLID的组成,前两个小写字母是目及以上分类地位的代表(历史原因脊椎动物这里只有一个字母),接着是属名的前三个字母(第一个字母大写),和种加词的前四个字母(第一个字母大写),最后加上一个代表采样个体的数字(这个数字是按这个物种的采样顺序给的),更完整的版本还会在数字之后加上.version代表基因组的组装版本。
- ToLID的一个例子,dhBauVari1.2。代表洋紫荆(Bauhinia variegata)物种的第一个个体的第二个组装版本。
- 其中d代表双子叶植物,h代表豆目,Bau是羊蹄甲属Bauhinia的前三个字母,Vari是种加词variegata的前四个字母,1代表第一个组装个体,.2代表第二个组装版本。
- 代表目及以上分类地位的前两个字母可以在https://gitlab.com/wtsi-grit/darwin-tree-of-life-sample-naming/-/tree/master/上的prefix_assignment.xlsx文件里查询到。
3. 评估指标和评估工具
参考:https://www.earthbiogenome.org/assembly-standards。
3.1. 评估指标
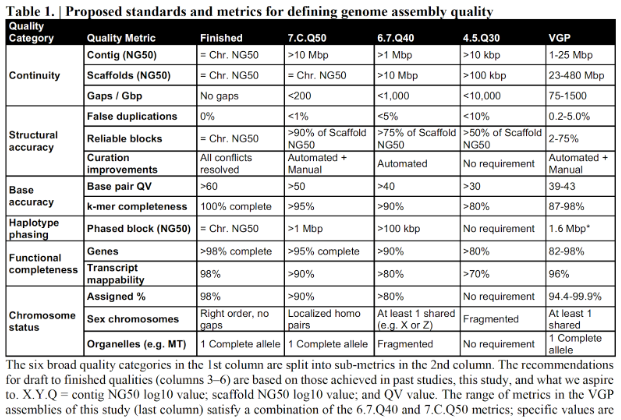
Figure 2. EBP建议的基因组组装质量评估指标 图源:https://www.earthbiogenome.org/assembly-standards
3.2. 评估工具

Figure 3. EBP建议的基因组组装质量评估工具 图源:https://www.earthbiogenome.org/assembly-standards
3.3. 其他
本人总结了几种个人常用的指标和工具,包括:
- QUAST:评估基因组scaffold N50,L50等指数。
- BUSCO:基因组和预测的蛋白组都可以用BUSCO评估基因组组装或注释的完整度(completeness)。
- LAI:通过LTR组装指数评估基因组的连贯性(continuity)。
- raw data mapping:把测序的reads(包括pacbio,Illumina,RNA-seq reads等)映射回组装好的基因组,评估mapping rate,genome coverage,depth分布等指标。这有非常多工具可用。
- 除了EBP建议的工具,还有许多其他工具。
- 比如FastaSeqStats用来评估基因组组装。
- 对于污染(contaminations),尤其是小的contigs的污染,还可以用blobtools进行评估和处理。
4. references

Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK