
0

使用SomaticSignatures进行突变数据的signature的判断
source link: https://www.bobobk.com/871.html
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

使用SomaticSignatures进行突变数据的signature的判断
2022年4月9日
|
技术
SomaticSignatures包于2015年发表在bioinformatcis杂志上,该杂志为专业的生物信息学杂志,该包旨在通过对肿瘤的single-nucleotide variants(SNP)数据进行分析,找到肿瘤发生发展,演化机制。本文将介绍如何使用snv数据分析得到该肿瘤的特征snp。
数据需要的是SNP数据,其中需要的数据有"Sample",“chr”, “pos”,“ref”, “alt”,分别为样本,染色体,snp起始位置,结束位置,参考ref的碱基,alt的碱基。对于下载到的TCGA的数据,我还是使用python处理好。 原始数据
import pandas as pd
df = pd.read_csv("TCGA.CRC.mutect.maf.csv")
df.head()
"""
Hugo_Symbol Entrez_Gene_Id ... MC3_Overlap GDC_Validation_Status
0 UBE2J2 118424 ... True Unknown
1 RPL22 6146 ... True Unknown
2 TNFRSF9 3604 ... True Unknown
3 EXOSC10 5394 ... False Unknown
4 PTCHD2 57540 ... True Unknown
"""
df = df[df.Variant_Type=='SNP']
#首先筛选snp数据
newdf = df.iloc[:,[33,4,5,11,12]]
#挑选特定列
newdf.columns = ["Sample","chr", "start","ref", "alt"]
newdf.ref = newdf.ref.str[:1]
# 去除ref与alt一样的行,否则后续会出错
newdf = newdf[newdf.ref != newdf.alt]
#保存数据用于R分析
newdf.to_csv("TCGA.CRC.mutect.maf.filtered.csv",index=None)
筛选Signatures
这里采用R来进行分析,以为要使用到SomaticSignatures包,首先安装必要的包
install.packages(c("SomaticSignatures","SomaticCancerAlterations","BSgenome.Hsapiens.UCSC.hg38","data.table"))
suppressPackageStartupMessages(library(SomaticSignatures))
suppressPackageStartupMessages(library(SomaticCancerAlterations))
suppressPackageStartupMessages(library(BSgenome.Hsapiens.UCSC.hg38))
suppressPackageStartupMessages(library(data.table))
suppressPackageStartupMessages(library(ggplot2))
读入数据并将数据导入成mutationContext需要的数据
df=fread('TCGA.CRC.mutect.maf.filtered.csv',data.table = F)
# ["Sample","chr", "start","end","ref", "alt"]
alls=as.character(unique(df$Sample))
df$study=df$Sample
sca_vr = VRanges(
seqnames = df$chr ,
ranges = IRanges(start = df$start,end = df$start+1),
ref = df$ref,
alt = df$alt,
sampleNames = as.character(df$Sample),
study=as.character(df$study))
运行mutationContext并将signature画出来并检查其表达的方差
sca_motifs = mutationContext(sca_vr, BSgenome.Hsapiens.UCSC.hg38)
head(sca_motifs)
# 对每个样本,计算 96 突变可能性的 比例分布情况
escc_sca_mm = motifMatrix(sca_motifs, group = "study", normalize = TRUE)
dim( escc_sca_mm )
table(colSums(escc_sca_mm))
head(escc_sca_mm[,1:4])
n_sigs = 5:15
gof_nmf = assessNumberSignatures(escc_sca_mm , n_sigs, nReplicates = 5)
save(gof_nmf,file = 'gof_nmf.Rdata')
load(file = 'gof_nmf.Rdata')
# 这个 assessNumberSignatures 步骤耗时很严重。
pdf("plotNumberSignatures.pdf",width=18, height=7)
plotNumberSignatures(gof_nmf)
dev.off()
结果显示
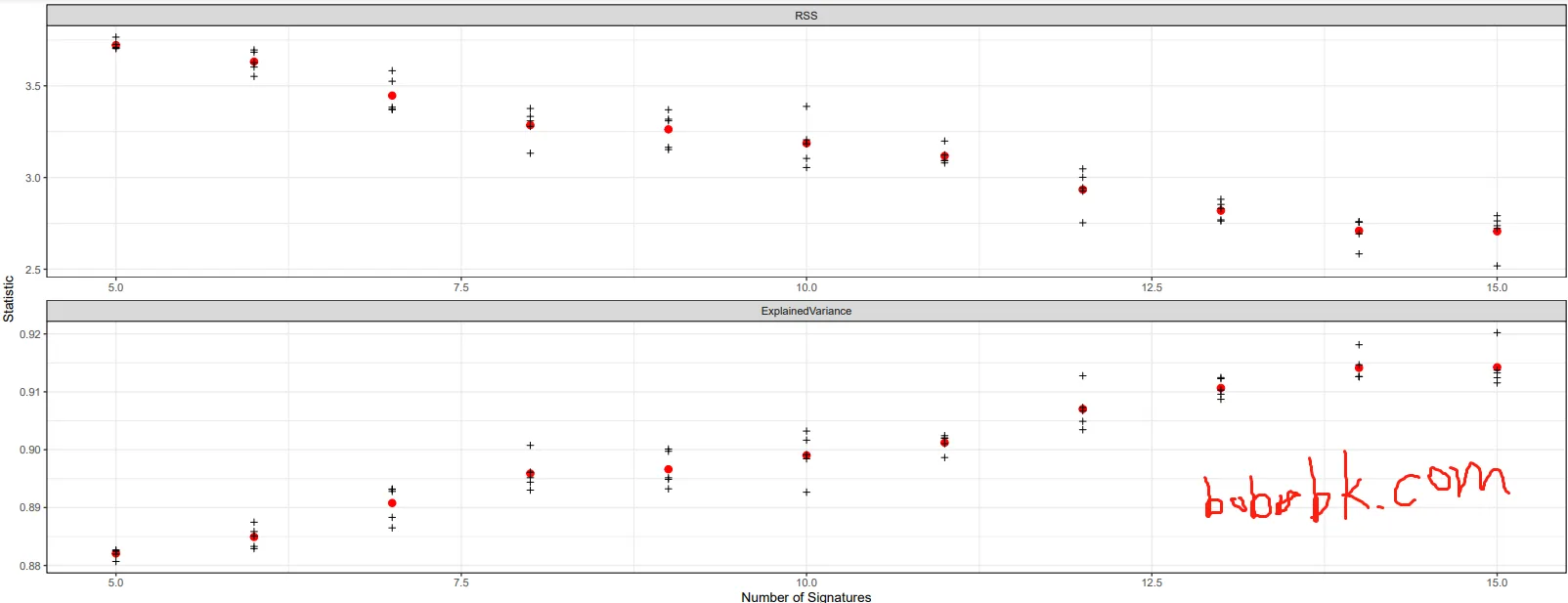 当signature达到13时便差不多到达99%的方差了
当signature达到13时便差不多到达99%的方差了
signature的绘制
最后我们通过前一步筛选的somatic muatiaon的signature
sigs_nmf = identifySignatures(escc_sca_mm ,
11, nmfDecomposition)
save(escc_sca_mm,sigs_nmf,file = 'escc_denovo_results.Rata')
load(file = 'escc_denovo_results.Rata')
str(sigs_nmf)
library(ggplot2)
pdf("maf.pdf",width=18, height=7)
plotSignatureMap(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Heatmap")
plotSignatures(sigs_nmf, normalize =T) +
ggtitle("Somatic Signatures: NMF - Barchart") +
facet_grid(signature ~ alteration,scales = "free_y")
dev.off()
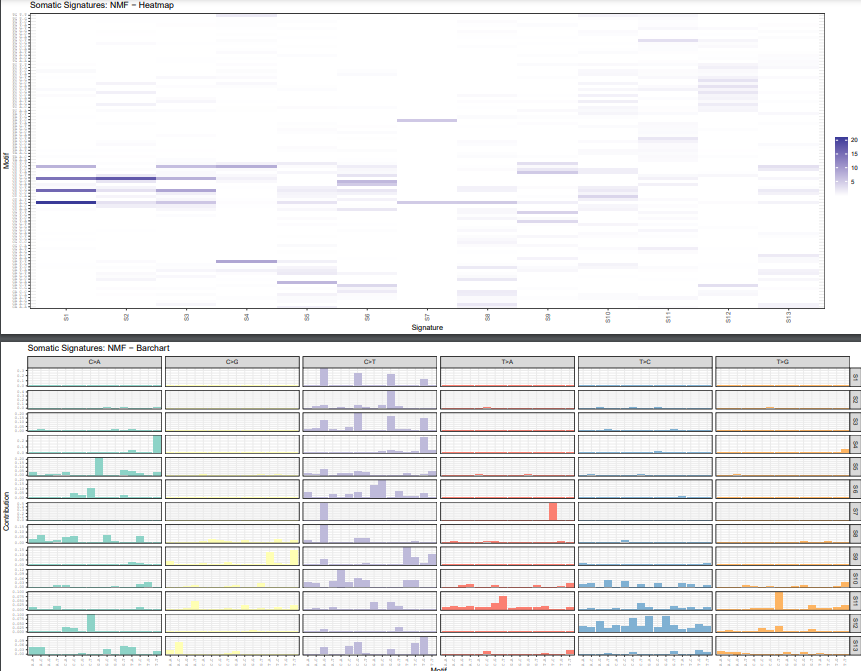
本文用于在肿瘤数据中筛选出需要的signature的snp(可以使用病人数据或者TCGA公共数据),而不是采用cosmic数据库的30个肿瘤突变signature,对于特定肿瘤方面的专家可以找到更d意d的结果。
See Also
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK