

Immunodominant T-cell epitopes from the SARS-CoV-2 spike antigen reveal robust p...
source link: https://www.nature.com/articles/s41598-021-92521-4
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.

Introduction
Uncovering the immunological responses to COVID-19 infection will help in designing and developing next-generation therapies and manage the treatment of critical COVID-19 patients. Many host factors associated with mild or severe disease symptoms have been reported. For example, leukopenia, exhausted CD8 T-cells, higher levels of TH2 cytokines in serum, a high titer of neutralizing antibodies, blunted interferon response, dysregulation of the myeloid cell compartment, activated NK cells, and the size of the naïve T-cell compartment is associated with critically ill patients1,2,3,4,5. This wide range of variable factors shares a common immunological underpinning—that of a systemic dysregulation in immune homeostasis due to the failure of the host immune system to clear the virus during the early stages of the infection6. Animal and human studies have demonstrated that susceptibility to respiratory virus infections is associated with compromised CD8 T-cell immunity7,8,9,10,11. A delay in the activation of CD8 T-cells and a lack of early IFN-γ production by the innate immune arm leads to an increase in viral load triggering overactivation of the innate and the adaptive arm of the immune system leading to a loss of immune homeostasis resulting in severe disease phenotype, including death. Therefore, an early wave of strong CD8 T-cell response may delay viral titer build-up, allowing rapid clearance of the virus by the immune system without perturbing immune homeostasis.
Healthy humans not exposed to COVID-19 show pre-existing CD4 and CD8 T-cell immunity to SARS-CoV-2 antigens12,13,14. The pre-existing immunity to CD4 and CD8 T-cells was detected against structural and non-structural SARS-CoV-2 proteins by overlapping 15-mer peptide pools. The existence of a pool of SARS-CoV-2-reactive T-cells in unexposed individuals is thought to arise from coronaviruses that cause common cold13,15,16. Whether pre-existing immunity provides any protection to SARS-CoV-2 infection or contributes to a faster recovery from infection remains speculative. Besides, it is unclear whether a pre-existing immunity, involving either CD4 or CD8 T-cells, or both, is required for maximal protection. Identifying robust pre-existing immunity against SARS-CoV-2 in the healthy population can be used as a measure to assess the mode of recovery and also viral spread in the global population.
In this study, we identified strong CD8 T-cell-activating epitopes from SARS-CoV-2 spike protein by a combination of epitope prediction and T-cell activation assays in healthy donors unexposed to SARS-CoV-2. The rationale for identifying epitopes that favor CD8 T-cell activation was twofold. First, robust CD8 T-cell activating epitopes can be formulated as second-generation vaccines for short and long-term protection against viral infection. Second, detection of pre-existing immunity in healthy donors using epitopes that favor CD8 T-cell activation may provide a framework to understand the complex immune responses observed in clinical settings. It may also shed light on the differences in morbidity and mortality in different population groups across the globe.
We developed a proprietary algorithm OncoPeptVAC to predict CD8 T-cell activating epitopes across the SARS-CoV-2 proteome. OncoPeptVAC predicts binding of the HLA-peptide complex to the T-cell receptor (TCR). We selected a cocktail of eleven 15-mer peptides with a broad class-I and class-II coverage and favorable TCR engagement predicted by the algorithm. The cocktail of peptides was tested for T-cell activation in healthy donors from the USA and India unexposed to COVID-19. We observed higher CD8 T-cell activation by the 11-peptide pool compared to the overlapping 15-mer peptide pools from the spike-S1 and S2 proteins. Homology analysis of the selected peptides with other coronavirus spike proteins indicated a lack of significant amino acid identity with any of the 11 peptides, suggesting engagement of one or more peptides in the pool to cross-reactive TCRs from other viruses, not particularly from a coronavirus. Bulk and single-cell TCR analysis revealed expanded clonotypes recognizing epitopes from CMV, Influenza-A, and other viruses to which most of us are exposed. Taken together, our findings support that strong pre-existing CD8 T-cell immunity in unexposed donors is contributed by cross-reactive TCRs from other viruses. Significantly, we discovered multiple immunodominant epitopes in our predicted pool of peptides that favored CD8 T-cell activation. Finally, we show that our cocktail of 11-peptides induced a robust immune response in convalescent patients demonstrating that these peptides are recognized by infected patients. Taken together, our study uncovered strong pre-existing CD8 T-cell immunity against SARS-CoV-2 using a small set of 11 epitopes that engaged cross-reactive TCRs recognizing epitopes from other viruses, not necessarily common cold viruses belonging to the coronavirus family as hypothesized by other studies. Additionally, our findings raise the possibility that many individuals carrying antigen-experienced T-cells against other viruses may be naturally protected against COVID-19 without prior SARS-CoV-2 infection.
RESULTS
Prediction of immunogenic epitopes favoring CD8 T-cell activation
A deep CNN model OncoPeptVAC was implemented to predict the immunogenicity of peptides based only on the peptide and HLA sequences. A total of 8870 immunogenic and non-immunogenic peptide-HLA pairs were obtained from the IEDB17. The BLOSUM encoding was used to represent the peptide and HLA molecules. The BLOSUM substitution scores encode evolutionary and physicochemical properties of the amino acids18. In addition, hydrophobicity indices and predicted HLA binding scores were also used to represent the peptide and HLA sequences.
OncoPeptVAC used the CNN model with multiple 2D convolutional layers combined with max-pooling to confirm the additive effect of different input features on the performance of the model. All the model versions were trained using fivefold cross-validation. The AUC of the final model was 0.87 based on a blind test dataset (Fig. 1A). The prediction algorithm showed a sensitivity of 0.64 and a specificity of 0.84 based on the score cut-off of 0.2 (Fig. 1B). By increasing the cut-off score to 0.5, the specificity could be further increased to 96.8 with a concomitant loss in sensitivity. OncoPeptVAC reduced the number of false positives significantly compared to the HLA-binding rank (Compare Fig. 1B,C) reducing the number of epitopes by 30% that needed to be screened in a T-cell activation assay to identify true immunogenic epitopes. For example, to identify 50% (119 out of 238) of the immunogenic peptide-HLA pairs present in the blind test dataset, 256 top peptide-HLA pairs from OncoPeptVAC prediction needed to be screened, compared to 753 top peptide-HLA pairs predicted by netMHCpan-4.0.
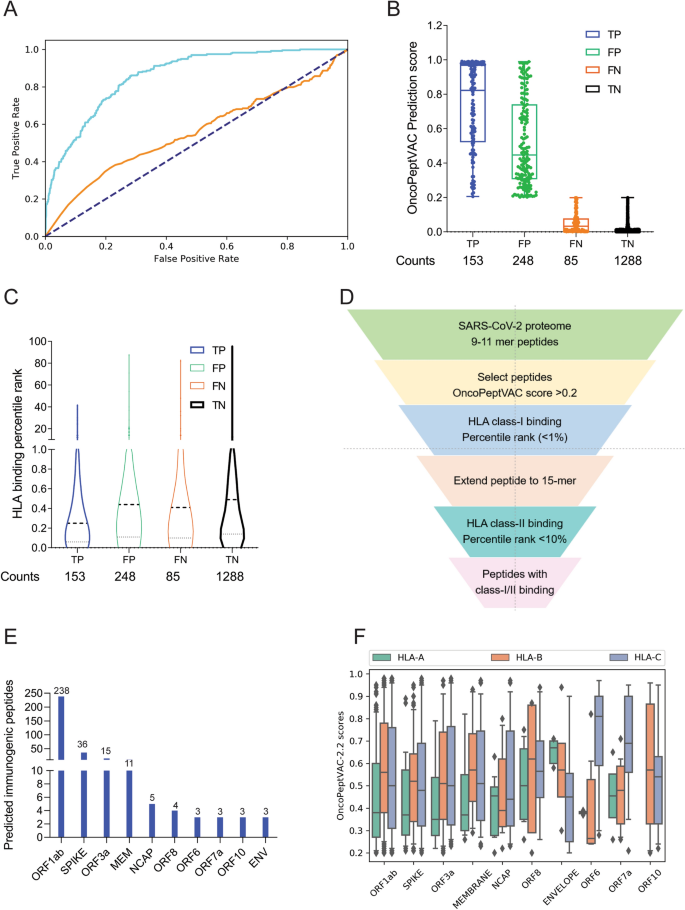
Identification of immunogenic epitopes from SARS-CoV-2 by OncoPeptVAC. (A) ROC curves of OncoPeptVAC TCR-binding and netMHCpan-4.1 HLA binding algorithms. A blind dataset of non-immunogenic or immunogenic HLA class-I binding T-cell epitopes from IEDB was used to assess the performance of OncoPeptVAC (cyan). The HLA-binding affinity of the epitopes expressed as percentile rank < 1% was used to assess the performance of netMHCpan-4.1 in predicting true immunogenic epitopes (orange). (B) Separation of immunogenic from non-immunogenic epitopes by OncoPeptVAC score. (C) Separation of immunogenic from non-immunogenic epitopes by HLA-binding percentile rank. (D) Schematic showing the steps used to identify immunogenic epitopes from SARS-CoV-2 proteome. (E) Number of immunogenic epitopes identified in different SARS-CoV-2 antigens. (F) HLA-A, B and C-restricted epitopes from SARS-CoV-2 proteome.
The prediction algorithm was applied to the SARS-CoV-2 proteome and screened against 23 class-I HLAs covering over 98% of the world population. A schematic of the in silico screening approach is shown in Fig. 1D. Briefly, 9–11-mer peptides from the SARS-CoV-2 proteome were screened for TCR-binding against 23 HLA, and peptides with OncoPeptVAC score > 0.2 were analyzed for class-I HLA binding. Peptide-HLA pairs with a high predicted binding affinity (< 1 percentile rank) were selected, their length extended to 15-mer, and screened for class-II HLA binding (See “Methods” for details). Peptides with favorable TCR binding and class-I/II HLA binding features were selected for further validation. The number of predicted immunogenic epitopes from SARS-CoV-2 protein-coding genes is shown in Fig. 1E. The distribution of OncoPeptVAC scores against different class-I HLA genes indicates a higher number of favorable TCR-binding peptides for HLA-B and C compared to HLA-A (Fig. 1F). Natural biases in HLA restrictions have been reported for immunogenic HIV epitopes19.
T-cells from unexposed donors respond to OncoPeptVAC-predicted peptides
We performed T-cell activation assay using a set of 11 prioritized epitopes from the SARS-CoV-2 spike antigen (Table 1) in unexposed donors. We selected epitopes from the spike antigen because it is highly immunogenic and generates strong B and T-cell responses. Besides, identifying strong immunogenic epitopes from the spike antigen can become useful reagents to study mechanisms of immune toxicity, and for long-term immune monitoring studies in naïve and vaccinated populations. The 15-mer peptides contained in the 11 selected epitopes cover different segments of the RBD and the non-RBD regions of the spike antigens and few peptides harbor ACE2 receptor binding sites (Fig. 2A and Table 1). Many of the predicted peptides reside in flexible regions of the spike protein that could favor efficient processing and presentation. Only two out of the 11 peptides showed 100% identity to the 315 peptides present in the spike-S1 and S2 pools obtained from a commercial vendor (Table 2) (See the “Methods” section for the creation of the peptide pool).
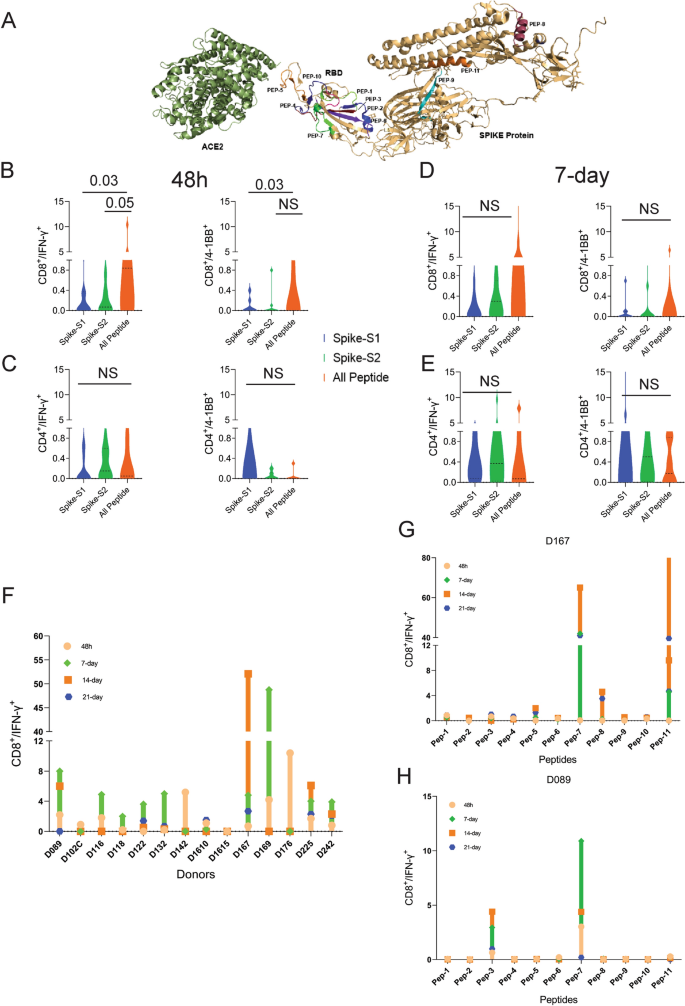
T-cell reactivity to SARS-CoV-2 Spike peptide pools and OncoPeptVAC prioritized peptides. Reactivity was determined by intracellular IFN-γ staining and surface expression of 4-1BB by FACS after stimulation of PBMCs from unexposed donors (n = 14) using separate pools of Spike-S1 and S2 peptides and the 11-Peptide-mix predicted by OncoPeptVAC (see “Methods”). (A) Structure of Spike—ACE2 receptor complex showing the location of the 11-peptides predicted by OncoPeptVAC. (B-C) T-cell activation after 48 h incubation with the peptides. (D-E) T-cell activation after a 7-day incubation with the peptides. (F) Kinetics and magnitude of CD8 T-cell activation in unexposed donor PBMCs. (G,H) CD8 T-cell response of donors D167 and D089 to individual peptides from the 11-peptide-mix. Statistical significance determined by Wilcoxon matched pairs signed rank test in Figures (B-E).
We screened PBMCs from 14 unexposed donors from the US collected between 2016–2018 and India (2015–2017), much before SARS-CoV-2 was recognized as a global pandemic (Table-S1). Activation of T-cells using the cocktail of 11 peptides (All-peptide mix) was compared to the responses from Spike-S1 (157 peptides) and S2 (158 peptides) pools (see “Methods” for assay details). At 48 h, the All peptide-mix induced a strong IFN-γ response in CD8 T-cells (Fig. 2B left panel), and a weaker response in CD4 T-cells (Fig. 2C, left panel). The IFN-γ response by the All peptide-mix was stronger in CD8 T-cells, compared to responses from spike-S1 and S2 peptide pools (Fig. 2B, left panel). The 4-1BB response in both cell types was weaker at 48 h (Fig. 2B,C right panels). IFN-γ levels increased in CD8 T-cells at day-7 by the All-peptide mix compared to the Spike peptide pools (Fig. 2C, left panel). The higher expression of IFN-γ and 4-1BB in the CD4 and CD8 T-cells in certain donors at 7-day suggested de novo activation (Fig. 2D,E). At 48 h, 70% of the unexposed donors showed > 0.5% IFN-γ response in CD8 T-cells to the predicted peptide mix suggesting recall to pre-existing antigen-experienced CD8 T-cells (Fig. 2F). In most donors, the maximal response was detected by 7-days, but in donors D142 and D176 the response peaked at 48 h and declined thereafter (Fig. 2F). Although the use of 15-mer peptides is expected to skew the response towards CD4 T-cells, we observed a stronger CD8 T-cell response to the Peptide-mix suggesting that the 15-mer peptide though added exogenously, was processed and presented by class-I HLAs efficiently. Taken together, the results demonstrate that the use of OncoPeptVAC identified potent CD8 T-cell epitopes in the spike antigen that could not have been detected by using large overlapping peptide pools used in T-cell activation assays.
Next, we tested individual peptides from the mix to assess their contribution to T-cell activation. The magnitude and kinetics of IFN-γ and 4-1BB induction in CD8 T-cells by individual peptides were variable in different donors (Fig. 2G,H and Figure S1, S2). Peptide-7 was an exceptionally strong CD8 T-cell epitope inducing IFN-γ in 3 out of 7 donors (Fig. 2G,H and Supplementary Figure S1E). Most immunogenic epitopes activated CD8 T-cells at 48 h and achieved > 2% activation by 7-day. The early activation at 48 h suggested that these epitopes engaged pre-existing T-cell immunity in the unexposed donors. However, activation at day-7 could come from both pre-existing and naïve T-cells. In many donors, a strong CD4 T-cell response was detected by individual peptides (Figure S3-4). Interestingly, in many donors, both IFN-γ and 4-1BB expression was induced by multiple individual peptides, although the magnitude of response by the All-peptide mix was not always additive (compare donor responses to the All peptide mix, Fig. 2F, with responses to individual peptides, Fig. 2G,H and Figure S1 for IFN-γ and Figure S2A with S2B-H for 4-1BB) confirming that the T-cell activation potential of individual peptides could be masked, when present as a part of a larger peptide pool.
Multiple studies have reported pre-existing T-cell immunity in unexposed donors using spike peptide pools and attributed the response to T-cells recognizing epitopes from common cold-causing coronaviruses to which a large section of the global population is exposed12,13,15. Homology analysis of the selected epitopes (see “Methods”) indicated that 6 out of the 11 peptides share > 67% sequence identity with SARS-CoV and only 1 (Peptide-11) out of the 11 peptides has over 70% identity with multiple coronaviruses (Table 3). Peptide-11 is in the S2 domain of the spike protein and showed ≥ 1% CD8 T-cell response at 48 h in 1 out of 7 donors tested (D167, Fig. 2G). However, peptides 3, 6, 7, and 9 lacking significant identity to other coronaviruses (Table 3) showed ≥ 1% CD8 T-cell activation at 48 h in at least one donor out of 7 (Fig. 2G,H and Figure S1). Peptide-7 induced high CD8 T-cell activation at 48 h in two donors (D089 and D225 (Fig. 2F and Figure S1E). Taken together, the data suggest that pre-existing T-cell immunity to these peptides may be derived from cross-reactive TCRs recognizing other viruses.
Analysis of antigen-specific CDR3s in responsive donors
To identify CDR3s amplified by individual peptides, or the All-peptide-mix, bulk TCR analysis was performed on antigen-stimulated PBMCs from donors D089 and D225 at days 7, 14, and 21 post-stimulation with antigens (see “Methods”). Both donors showed a robust IFN-γ response to Pep-7, and the Pep-Mix, but not to Pep-1 (Figure S4). Diversity and clonal amplification of unique public and private CDR3s were analyzed at three different time points (Fig. 3A,B). Both the donors showed clonal expansion of multiple public CDR3s recognizing HCMV, human herpes virus-5 (HHV-5), and Influenza-A peptides when stimulated with Pep-7 and All-peptide mix, but not with Pep-1 (Table S2). HCMV and HHV-5 CDR3s were expanded in donor D089 (Fig. 3A, top panel), whereas D225 showed expansion of HCMV and Influenza-A CDR3s (Fig. 3A, bottom panel). Significantly, these CDR3s were not amplified by Spike-S1 and S2 peptide pools or by Pep-1, the latter failed to activate T-cells in these donors. Further, CDR3s recognizing HCMV peptide NLVPMVATV in donors 089 and 225 were different, suggesting that the same antigen engages multiple cross-reactive TCRs in different donors. Next, we analyzed private CDR3s in these two donors to identify novel SARS-CoV-2 antigen-specific CDR3 (Fig. 3B). Donor 089 showed a lack of specific amplification of private CDR3s suggesting that the robust CD8 T-cell response detected in this donor may be contributed by the amplified public CDR3s (Fig. 3B, top panel). In contrast, two private CDR3s were clonally amplified by Pep-7 and All-peptide mix in D225 suggesting that the T-cell response is derived from both public and non-public TCRs in this donor (Fig. 3B, bottom panel). Given that the TCR analysis was performed on day-7 and beyond, the amplified CDR3s may be derived from both pre-existing and naïve T-cells. A list of clonally amplified public and private CDR3s detected in the two donors is given in Table S2.
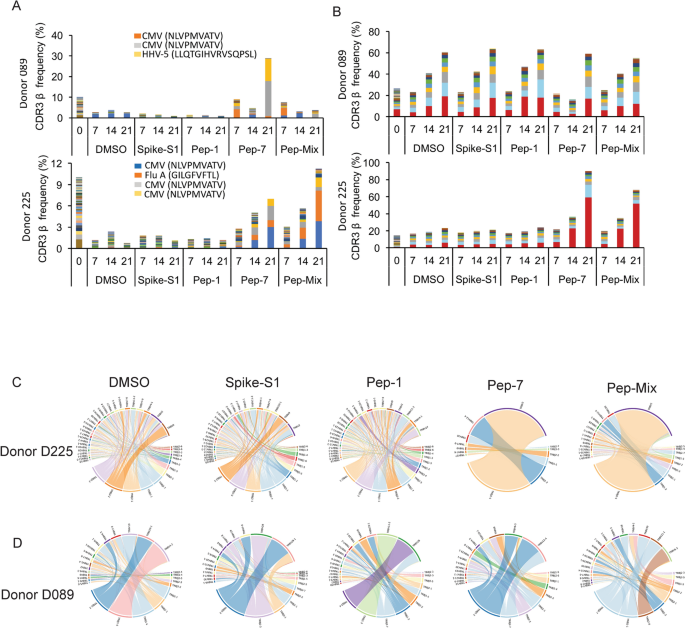
Bulk TCR repertoire analysis after in unexposed donors following in vitro stimulation of PBMCs at different time points with the indicated peptides. (A) Expanded public CDR3-βs recognizing shared antigens in the D089 (upper panel) and D225 (lower panel). (B) Expanded private CDR3-βs in D089 (upper panel) and D225 (lower panel). (C) V–J gene usage in D225. (D) V–J gene usage in D089.
To further investigate the TCR repertoire profile of donors 089 and 225 we analyzed the VDJ gene usage in the bulk CDR3 data. In D225, two V segments TRVB2 and TRVB30, and a J gene TRBJ2-1 were significantly over-represented in Pep7 and Peptide-mix treated samples (Fig. 3C), whereas in D089 TRBV12-4 and TRBJ1-2 genes were amplified (Fig. 3D).
Single-cell gene expression and TCR profiling of activated T-cells
To characterize the phenotype and functional state of activated T-cells and reveal differences between the different treatments, we performed single-cell sequencing on a 10X platform. Single-cell transcriptomics and TCR data obtained from 3500–4500 cells identified 3000–3500 unique transcripts (see “Methods”). Using graph-based clustering of uniform manifold approximation and projection (UMAP) we captured transcriptomes of 4 distinct cell types (Fig. 4A and Table S3). Our assay method is enriched for the growth and proliferation of T-cells causing depletion of other immune cell types present in PBMC in a 14-day culture. Three cell types, CD8, γ/δ, and NK-T were detected in all the samples. Compared to DMSO and Pep-7 in which the CD8 T-cell fraction was ~ 60%, in spike-S1 and spike-S2 the CD8 T-cell fraction was 50% and 38% respectively. Conversely, the CD4 cluster was expanded in spike-S1 (12%) and S2 (27%) compared to DMSO (7%) suggesting that the spike peptide pools engaged CD4 T-cells (Table S3). The single-cell transcriptomic analysis further revealed that Pep-7 induced effector phenotype in the CD8 T-cell cluster by the expression of activation markers IFN-γ, 4-1BB (Fig. 4B) TNFRSF9, FAS, and TIGIT (Compare Figure S5D with S5A-C). The top 10 Pep-7-expanded clonotypes were CD27+/SELL- suggesting transition towards effector memory phenotype (Fig. 4B). Spike-S1 and S2 peptides induced CD27+/SELL- T-cells in the CD4 clones 6 and 4 respectively (Fig. 4B). Single-cell data revealed amplification of TRBV2 (40%) and TRBJ2-1 (32%) in Pep-7 stimulated T-cells (Fig. 4D) confirming the results from the bulk TCR analysis (Fig. 3C,D).
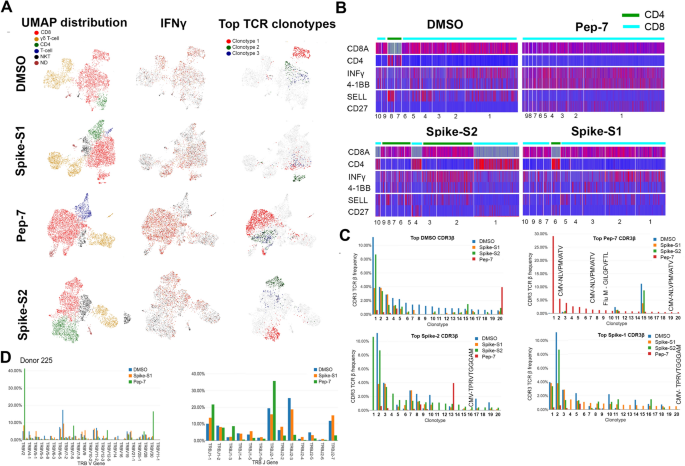
UMAP projection of different cell types identified in unexposed donor D225 after 14 days of in vitro stimulation assay with different antigens. (A) Clusters of different cell types and their relative proportions present in the assay mixture (left panel). Clusters expressing IFN-γ (middle panel) and the top-3 amplified clonotypes (right panel). (B) Heat map showing the expression of cell-type and cell-phenotype-specific markers in the top-10 amplified TCR-β clones. (C) Frequency of CDR3-β recognizing public and private antigens in the top 20 clonotypes. (D) Amplified V and J-genes in the top-20 clonotypes.
Next, we mapped CDR3-β to specific clones from each treatment (Fig. 4C). The DMSO, spike-S1, and S2-treated samples shared many clonotypes among themselves in the same frequency range suggesting weak antigen-induced activation and proliferation of T-cells. However, Pep-7 treated sample was enriched in many CDR3-β clones absent in other samples indicating the specificity of response (Fig. 4C, red bars). Four clones among the top-20 clones encoded CMV and flu-specific CDR3-βs in Pep-7 treated sample but not in other samples confirming the findings from the bulk TCR data that the peptide engaged cross-reactive TCRs (Fig. 4C). The expanded CDR3-β detected in the Spike-S2 treated sample belonged to CD4 T-cells (Fig. 4B, Spike-S2 panel, clonotype-1). Taken together, single-cell TCR analysis demonstrated that the immunogenic SARS-CoV-2 epitope engaged many unique CDR3-βs not shared by spike-1 and spike-2 peptide pools.
The bulk and the single-cell TCR analyses demonstrated that the SARS-CoV-2 epitope identified in this study engaged both cross-reactive public CDR3s and unique CDR3s not associated with known viral antigens and favored specific V-J gene usage. Further, the expansion of TRBV-2 and TRBJ2.1 by Pep-7 and by the Pep-mix in D225 confirmed that out of the 11-peptides contained in the Pep-mix, Pep-7 contributed to all of the T-cell responses observed in this donor.
Antigen-specific clonal expansion and T-cell phenotype
Next, we analyzed the clonal composition and phenotype of T-cells to investigate the dynamics of antigen-specific T-cell response in the treated samples. We analyzed the top-30 clones for their phenotype by the expression of 25 marker genes (Figure S6). In all samples, including DMSO, CD8 T-cell clonotypes were more frequent (Figure S6A-D). As expected, the CD4 T-cell compartment was expanded in Spike-S1 and S2-treated samples (20 and 25% of all clonotypes respectively) compared to DMSO (6.5%) (Figure S6B-C). The CD4 T-cells expressed TNFSF4 (OX-40) suggesting activation, although they failed to express IFN-γ (Figure S6B-C). A few expanded CD4 clones in the Spike-S1 and S2 treated samples showed a high expression of IL17RB suggesting polarization towards a TH17 phenotype (Figure S6B-C). In the Pep-7 treated sample, almost all clonotypes in the top-30 were CD8 T-cells. The highly expanded clones expressed multiple T-cell activation markers (Figure S6D). Interestingly, in addition to the activation markers, these cells expressed higher levels of IL2RA (CD25) suggesting differentiation towards an effector memory phenotype (Figure S6D). CD25 expression was low in the CD8 T-cell compartment in other samples. Taken together, the results of the transcriptomic analysis highlighted that the strong immunogenic CD8 T-cell epitope identified in this study preferentially engaged CD8 T-cells pushing them towards an effector and effector memory phenotype. The spike-S1 and S2 peptide pools on the other hand engaged both CD8 and CD4 T-cells and modulated the CD4 T-cells towards a TH17 phenotype.
Response of convalescent COVID-19 patients to predicted epitopes
To assess whether the predicted epitopes are recognized by COVID-19 infected patient T-cells, we tested the spike-S1, S2, and All-peptide mix on seven asymptomatic, five with mild-moderate symptoms, and five severe convalescent patients requiring ICU admission (Table S4) and analyzed their CD4 and CD8 T-cell response after 48 h. There was a significant donor to donor variation in T-cell response to all the antigens, however, patients experiencing mild to moderate symptoms exhibited slightly higher induction of IFN-γ in CD8 T-cells (Fig. 5A,B and Figure S13). Asymptomatic (AS) and severe patients showed weaker CD8 and CD4 T-cell response to all antigens (Fig. 5) Spike-S2 peptide pool induced stronger 4-1BB induction in CD8 and CD4 T-cells compared to the All-peptide mix (Fig. 5B,D). Taken together, our results confirm that the epitopes prioritized by the algorithm were recognized by COVID-19 infected patient T-cells, and the IFN-γ response induced by the All-peptide mix was skewed towards CD8 T-cells in line with our assay results on unexposed donors (Figure S13).
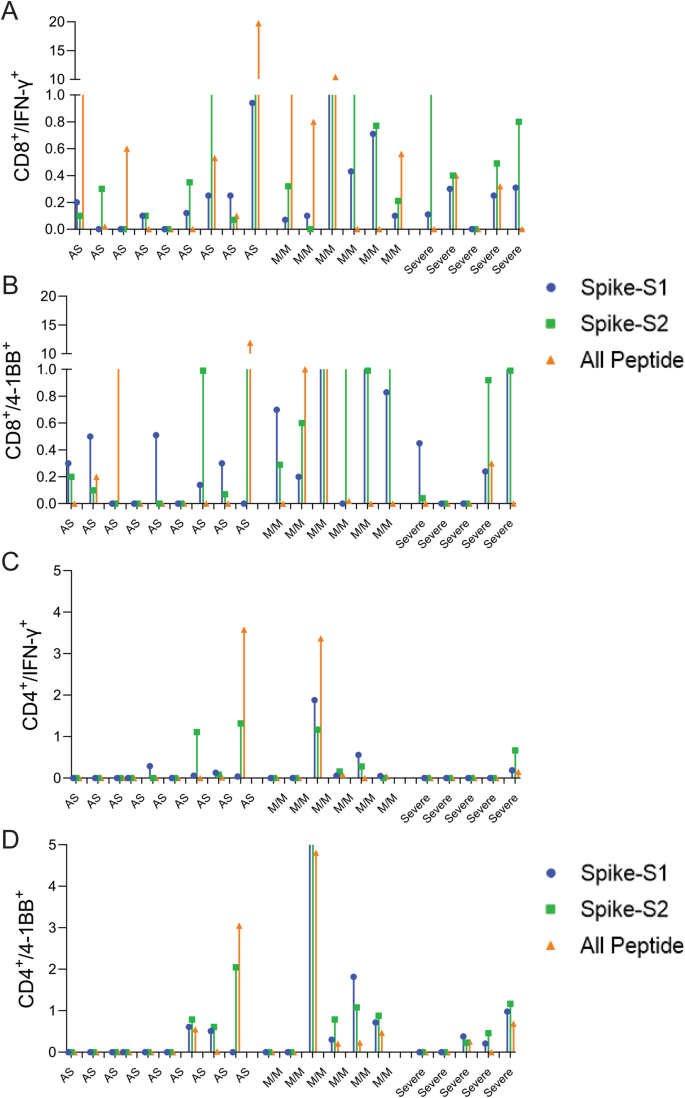
T-cell reactivity to Spike-S1, S2 pools, and 11-peptide-mix in asymptomatic, mild-moderate, and severe disease patients after in vitro stimulation for 48 h. (A) Convalescent patient PBMC collected between 45–60 days after PCR confirmation of COVID-19 infection was incubated with spike-S1 or S2 peptide pools or the All-peptide mix. T-cell activation was quantitated by intracellular IFN-γ and 4-1BB expression by FACS. T-cell activation for individual donors is plotted. (A,B) IFN-γ and 4-1BB expression in activated CD8 T-cells in asymptomatic (AS, 9 patients), mild/moderate (M/M, 6 patients) and severe (5 patients). (C,D) IFN-γ and 4-1BB expression in activated CD4 T-cells in the three patient groups. Differences in T-cell activation between groups were not statistically significant by Students T-test. Group-wise aggregated data is shown in Figure S13.
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK